作者: 姜翰昕 郭錦龍 / 臺灣大學材料系 博士 副教授
在前一篇電子報中,我們介紹了VASP計算的基礎設定,也強調了交換相關能選擇的重要性。在這份電子報中,我們將簡介如何使用VASP計算系統的能量態密度(density of states, DOS)與能帶結構。這兩者皆為材料的第一原理計算中最基礎的操作,計算時用到的技巧也是日後若需要進行更複雜計算的基礎。
如同上一份電子報,在這份電子報中提到的指令皆可在VASP wiki上找到更詳盡的說明[1]。
一、矽之能量態密度(density of states)計算
能量態密度(density of states, DOS)的定義為在單位能量中所擁有的本徵值能態數量與能量的關係。藉由能量態密度的計算,我們可以判斷系統為導體或是絕緣體。此外,如果我們進一步透過將Kohn-Sham軌道投影的方式將能態密度進行投影,也可看出系統中特定本徵值能量的能態主要是來自何種原子的貢獻。在材料設計上,這些資訊可做為如何透過摻雜改善系統的光電性質的重要參考。

進行矽的能量態密度計算之前,我們必須先建立結構已經完全被最佳化的矽製之primitive cell,而建立過程可參閱前回之電子報內容。與上次不同的是,這份電子報內所有計算的exchange-correlation function都是使用PBE(Phys Rev Lett. 1996 Oct 28;77(18):3865-3868.)而非PW-91。當我們使用VASP將結構已最佳化的矽之primitive cell進行單點計算(NSW=0, IBRION=1)時,DOS的資訊可被直接輸出至名為DOSCAR的輸出檔。若要畫出好看的DOS,以下指令扮演重要角色:
在以上指令中,ENMAX和ENMIN分別為計算DOS時的能態能量上下限,而NEDOS則是在計算中將此能量範圍分割的格點數。亦即,若我們打上以上指令,將會輸出本徵值能量範圍在-25 ~ +25 eV的結果,而整段能量範圍切割為250點,意即每個格點代表之能量範圍為0.2 eV。ISMEAR與SIGMA則是控制DOS之平滑度。由於電子能量態是離散的,故如果我們直接以長條圖的形式進行作圖,其圖中的曲線相當離散且難以與文獻結果進行比較。在這些設定中,我們將離散的各個本徵值能態轉化為高斯函數(ISMEAR = 0之意涵),且其寬度為0.01 eV (SIGMA = 0.05)。如此一來,我們即可得到較為平滑的曲線。

除了INCAR中設定,KPOINTS檔案中定義的k-point在倒晶格內的取樣方式也是此部分計算的重要關鍵。其影響在後面段落中將會更進一步提到。在此,我們使用Monkhorst-Pack的方式將倒晶格空間切成10×10×10的網格,而其設定如下:

當完成SCF計算後,我們可以從輸出的DOSCAR檔中擷取我們需要的部分內容:
若欲得到DOS的相關資訊,我們可以直接由第六行開始讀起。第六行中的第三與第四個數字分別為NEDOS的值以及根據我們計算條件所得出的Fermi level (Ef)。第七行之後的數字則是DOS作圖直接相關的資訊,這三列數字依序為能量區間的中點、電子DOS、以及DOS至此能量區間之積分值。(若在計算中有將spin-polarization打開,輸出的檔案將總共含有五列數字,依序為能量區間中點、spin-up電子對應的DOS、spin-down電子對應的DOS、spin-up DOS的積分值、以及spin-down DOS的積分值。)我們將這些資料作圖,即可得到如下圖的能量態密度與能量的關係:

根據上圖,我們可以看到矽的DOS曲線中在E - Ef = 0上方有個小小的不連續區。此不連續區即為能隙(band gap)。在能隙之上為矽之導帶(conduction band)、之下為價帶(valance band)。根據此圖,矽的能隙值約為0.6 eV,而此結果與文獻中運用DFT求出的值相符。然而,此值其實遠小於實驗值的1.1eV。像這樣的能隙低估主要是由於DFT本身理論上的限制。若要得到與實驗值相符的能帶值,我們則必須要對計算結果進行修正。修正的方法包含使用scissor operator直接將能帶的寬度增加,或是在一開始進行計算時即使用如hybrid DFT等更高階的計算方法。
K-Point取樣與DOS作圖品質之關係檢證
在上文中,我們有提到k-point的取樣會影響到DOS的品質,在此我們將進行一個簡單的驗證。在此計算中,我們使用完全結構最佳化後之Si的primitive cell,並比較使用2×2×2、4×4×4、6×6×6、以及10×10×10的Monkhorst-Pack格點取樣後所作出的DOS。我們做出來的結果如下圖。從圖中可以很清楚的看到,k-point網格至少要取到6×6×6才有可能做出比較好的結果,而這也是實際作為論文中作圖時必須要考慮的問題。

二、石英(SiO2)之能量態密度計算
在上段中,我們有提到能量態密度的投影對於材料設計方式的重要性。在此,我們使用石英(α-二氧化矽)為例,計算在系統中氧與矽各自的能量態密度。
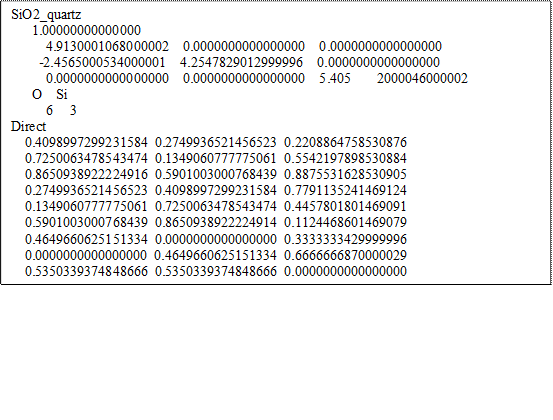
下面為經過結構最適化的石英primitive cell的POSCAR:

α-SiO2之primitive cell。圖中藍色原子為矽、紅色為氧。
在計算投影態密度時,我們只要在INCAR中加入”LORBIT = 10”之指令,在DOSCAR內即會將投影於各原子上的DOS以及其在不同軌域上之分量輸出於DOSCAR檔案中。由於此檔案中包含了系統的總DOS與投影至各原子的DOS,因此為一相當龐大的檔案。其檔案結構大致如下(在此計算中,由於系統內含有氧原子,所以我們有將spin polarization打開,即在POSCAR內設定ISPIN= 2):




在各原子的DOS中,我們可以看到總共有七列數字,從左自右分別為能階值、s軌域spin-up之分量、s軌域spin-down之分量、p軌域spin-up之分量、p軌域spin-down之分量、d軌域spin-up之分量、以及d軌域spin-down之分量。根據這些,我們可將DOS之資料收集並作圖,其結果如下(ISMEAR = 0.1):

此外,更進一步分析能量態密度在不同原子與軌域上可發現在valance band頂部的能態主要是由氧的p軌域所貢獻,而在conduction band底端之能態則為矽和氧的混成。
三、矽的能帶結構
除了能量態密度外,晶體結構的能帶結構(band structure)也是判斷系統光電性質相當重要的依據。能帶結構的定義為在周期系性統中電子的能量與動量間的關係,而根據能帶結構我們可推估系統的光電性質。下圖為文獻中Si primitive cell的能帶結構:
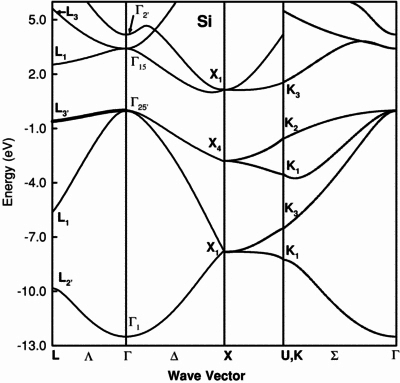
(Source:O Zitouni et. al. 2005 Semicond. Sci. Technol. 20 908)
我們也可藉由VASP計算將此圖重製。進行此計算時,我們首先將在2-1中完成結構最適化的Si primitive cell的CONTCAR複製為新的POSCAR,而POTCAR檔案則是使用與2-1相同的PAW-GGA(PW91)。同時,我們也在INCAR中加入LORBIT = 10之參數以輸出PROCAR檔案,做為能帶結構作圖所用。
最後則是KPOINTS檔的設定。進行能帶結構計算時,由於k-point取樣方式需要沿著特定方向,故無法使用如同前面計算所使用的網格式切法。因此,我們採用「線性取樣」法,沿著Brillouin zone中的高對稱方向並進行採點。囿於篇幅,在此無法完整介紹高對稱方向的決定方式,而只將我們使用的KPOINTS檔案列出。在此檔案中,我們選取四條Brillouin zone內的向量進行計算分析:L(0.5,0.5,0.5)-G(0,0,0); G(0,0,0)-X(0,0.5,0.5); X(0,0.5,0.5)-U(0.25,0.625,0.625); K(0.375,0.75,0.375)-G(0,0,0)。其檔案如下:

在此KPOINTS檔案中,第二行的”10”,代表每條路徑分割為10個k點,第三行的”line”表示此KPOINTS檔內定義的k-point為線性分割,而第四行的”reciprocal”則是表示下方各點的座標為倒空間(reciprocal space)中的相對座標。
此部分的計算結果可在”EIGENVAL”或”PROCAR”檔案中得到。以下將簡單介紹此二檔案之格式。
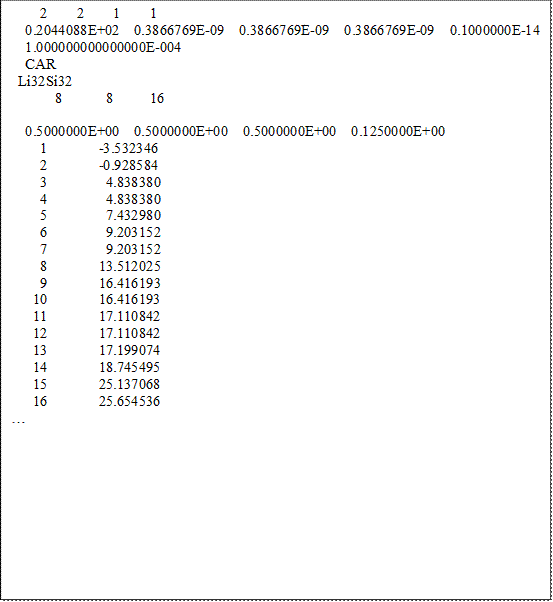
EIGENVAL
在此檔案中,前幾行為與本次計算相關的參數設定,而主要的內容是從第8行的”0.5 0.5 0.5 0.025”開始。此行的內容依序為k-point的x, y, 與z座標,以及此k-point在計算中所佔的權重(weight)。此行下面的數字則是代表各能帶的能量,依序為能帶編號、spin-up state的能階、與spin-down state的能階。由於在此計算中VASP預設是輸出16條能帶的能階值,因此我們可以看到1~16的數字。若想更改輸出的能帶數,可在計算時於INCAR中加入NEDOS = (能帶數)的參數設定。因為我們在KPOINTS中指定了40個不同的k-point,所以如果將檔案繼續往下拉可以看到與此區塊相同的文字結構重複40次。
將各k-point與對應的能階值輸出做圖,即可得到能帶結構。下圖為此系統中spin-up電子的能帶結構,而圖中E = 0點已被移至Fermi-level (Ef)。此圖之趨勢與能階值約與文獻值相符。

從能帶圖中,我們可以判斷出幾個重要的性質。首先,我們看到第二至四條能帶在G點上有明顯的重合,代表此系統在該點上有degenerate的情形。另外,此圖也顯示矽晶體的能隙值約為0.6eV而且是非直接能隙(indirect band gap)材料。
PROCAR
當在INCAR中加入LORBIT = 10的指令時即可輸出PROCAR。在這個檔案中除了有各k-point上對應之能階值外,還包含每個原子以及s、p、d等軌域投影於該能階的分量。此檔案的部分內容如下圖:

如同EIGENVAL,我們也可使用這部分的資料畫出band structure,得到與使用EIGENVAL內的資料作出的圖相同的結果。
四、結語
在本次的電子報中,我們簡單的介紹了如何使用VASP進行最基本的電子性質計算,而這些計算也是能透過DFT所執行的最基本計算。然而,VASP可以處理的計算還遠不只如此。在下次的電子報中,我們將會更進一步介紹如何使用VASP處理材料的光學與振動性質。
參考資料
[1]The VASP Manual: https://cms.mpi.univie.ac.at/wiki/index.php/The_VASP_Manual